New aspects of electrophylic aromatic substitution mechanism: Computational model of nitration reaction
Gas Phase
Figure 1 shows possible reaction stages, and those stationary points were subjects of our computational investigation. A particular feature of our results of gas-phase calculations was detection of a loose prereaction complex NO2+...C6H6. The vast area of PES with constant energy corresponds to this state. Located over aromatic ring is NO2+, at a distance of 3.3–3.5 Å without any preferences in orientation. It seems that this complex must be classified as solvate complex. Speaking in favor of this assumption is the fact that nitronium coordinates with O-containing benzene derivatives (-OCH3, -COOH, -NO2 etc) near the oxygen atoms, not the π-systems. In such cases, the choice of conformation of prereaction complex is ambiguous, so all enthalpies presented below are calculated relatively for the heat of formation of isolated reagents. The only inconvenience appears in cases of reactive substrates when effective activation barriers get formal negative value*. On the other hand, it is experimental fact that such gas-phase reactions can have negative temperature dependence of rate constants [9].
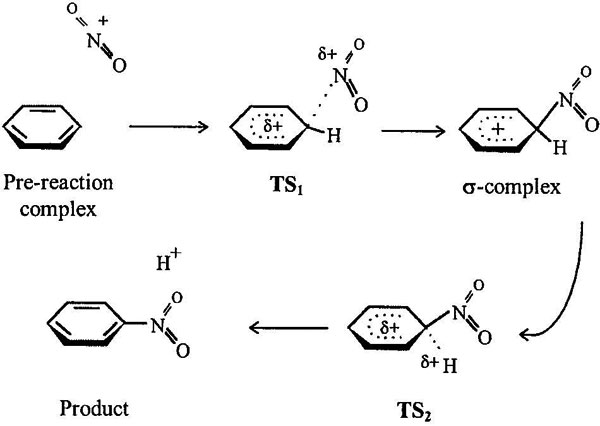 Figure 1. Possible stationary points on PES.
Figure 1. Possible stationary points on PES.
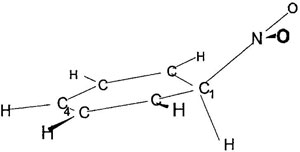 Figure 2. Geometry of σ-complex.
Figure 2. Geometry of σ-complex.
This “solvate” complex could be associated with existing in organic chemistry conception of π-complex if it were not a result of gas-phase calculations. Running a couple of steps ahead, there are no prereaction complex in liquid-phase calculations. In that case, the reagents are already solvated (see “Liquid Phase” section, below).
σ-Complex
Classic conception of aromatic nitration reaction includes interaction of positively charged N-atom of nitronium with π-orbital of carbon atom of benzoic ring. This reaction's direction leads to adduct, so-called “Wheland complex” or “sigma-complex” [3]. Our calculations gave its C−N distance to be 1.56 Å. Benzoic ring was slightly deformed, dihedral angles being 4–8° (see Figure 2). Judging by geometry of reactive center, it has a state somewhere between sp3 and sp2 hybridization. The C−H bond is a bit slack (it is lengthened by 4%, and the frequency of its valent vibration is 2800 sm−1 compared with ≈3100 of the rest C−H vibrations). Considerable redistribution of positive charge between nitrogroup and substrate occurs in this complex. Positive charge is delocalized between H-atoms (0.12–0.08, depending on the distance from reactive center) and reacting carbon atom (0.3); nitrogroup becomes practically neutral.
Further transformation, deprotonation, is not thermodynamically profitable in gas phase and needs nucleophil to form final product, nitrocompound. There exists an isomer of σ-complex, O-protonated nitrobenzene. Thermodynamically it is more stable, by 30 kcal/mol, but synchronous mechanism of NO2+ addition and proton transfer demands strained geometry of TS (rectangle) with distances of C...H and O...H being considerably longer than corresponding bond lengths.
Introduction of substituents into benzoic ring leads to appearance of four possible directions of reaction: para, meta, ortho, and ipso. Ipso attack (attack on substituted C-atom) was studied to evaluate the possibility of existence of ipso-σ-complex as an intermediate product. Ortho direction has its own particular feature: coplanar orientation of NO2+ becomes impossible because of steric interaction (spacious groups such as tret-butyl require entirely perpendicular orientation of planes of reagents). This should lead not only to increase of activation barrier but also to decrease of entropy of activation (and decrease of rate constants).
PES and reaction path
Formation of the σ-complex is preceded by TS. Here we discuss possible variants of TS geometry. There is common opinion that electrophils give π-complexes with aromatic compounds, which assumes their coordination over benzoic ring or one of its π-bonds. To investigate such possibility and to consider prereaction selectivity, the PES sections were calculated. Those sections describe changes of supermolecule's energy with nitronium movement at 3 Å over benzoic ring in the parallel plane** the distance of 3 Å corresponding to the initial part of reaction path (compare: prereaction complex, 3.5; TS, 2 Å). Such PES section of reaction with benzene looks like a hill with hollows along C-H bonds (Fig. 3). So central attack meets no attraction but a force pushing the electrophil to nearest C-atom. Thus we did not found anything as three-center TSs, possibilities of which were announced in Ref. [10].
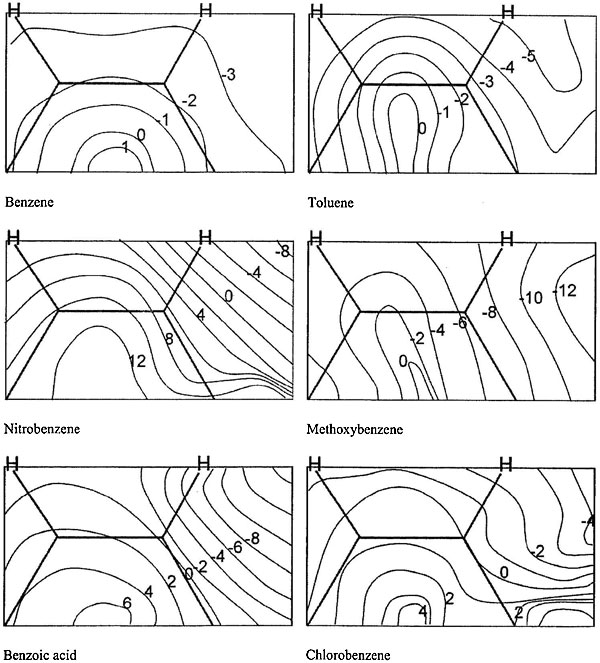 Figure 3. Sections of PES describing the movement of
Figure 3. Sections of PES describing the movement of NO2+ over aromatic ring at 3 Å. Energy values are shown relative to enthalpy of formation of reagents (kcal/mol). Geometries of reagents and their orientation are optimized at every point. Substituent is located at rightmost C-atom.
Presence of a substituent in aromatic ring deforms and shifts this hill from the center. In nearly all cases there is reduction of potential energy in the area of substituent caused by electrostatic attraction. An especially evident effect exists in cases of O-containing substituents that may serve the additional factor of ortho orientation.
A special subject pertaining to reaction path is the possibility of electron transfer. There is opinion based on experimental facts that NO2+ takes away an electron from substrate in pre-reaction phase, and thus formed ion-radical pair recombines in the next step of reaction, giving final products of nitration [8]. Let's tell the difference between two cases: transfer of electron density and electron transfer (ET). To avoid possible confusion, the first variant of interaction will be called “redistribution of electron density.” Indicator of the second variant is not a great change of atomic charges but appearance of biradical character of supermolecule. This issue is discussed below in the section titled “Charge and Electron Transfer.”
Transition states (TS)
The authors of Ref. [10] (3-21G, MP4) did not find any TS of reaction of nitronium with benzene. Quite the contrary, our calculations give TSs for nearly all substrates with the following characteristics: angle of attack of the ring ≈120°, C−N distance 1.97–2.22 Å. Benzoic ring keeps its planar conformation. Nitrogroup is bent at 145°. The TS is achieved both in coplanar and perpendicular orientation of reagents' planes, corresponding activation barriers differing less than 1 kcal/mol, excluding cases of ortho attack. Pseudo-vibration along reaction coordinate is practically pure NO2+ translation; no contribution of C-H vibration is discovered. This means that the TS of nitration in gas phase is “early”***. All these results are summarized in Table III, where geometrical and thermochemical characteristics of main stationary points on reaction path are presented.
C1-N | ∠C4C1N | C1-H | ∠C4C1H | qNO2 | qN | qH | qC6H5 | ΔΔHf | |
|---|---|---|---|---|---|---|---|---|---|
| Isolated reagents | ∞ | — | 1.100 | 180 | 1.000 | 0.679 | 0.130 | −0.130 | 0 |
| Prereaction complex | 3.3–3.5 | — | ∼1.10 | ∼180 | 1.000 | 0.684 | 0.155 | −0.155 | −6.1 |
| TS1 | 2.071 | 118 | 1.115 | 151 | 0.452 | 0.609 | 0.202 | 0.347 | 3.9 |
| σ-complex | 1.559 | 144 | 1.156 | 112 | 0.026 | 0.564 | 0.252 | 0.723 | −9.3 |
| Product | 1.487 | 180 | ∞ | — | −0.150 | 0.567 | 1.000 | 0.150 | 97.1 |
It was also found that there exists a possibility of reaction between protonated substrate and the molecule of neutral nitric acid: H-Ar-H+ + HO-NO2 ⇔ [H-Ar-NO2]+ + H2O. Calculations show that this reaction has practically the same activation barrier, although it demands proper orientation of reagents at synchronous H2O elimination: N-OH bond of nitric acid must settle itself over HC-CH2 bond of the benzoic ring. We must mention the alternative variant of reaction with nitronium: attack by oxygen atom. This pathway has higher (≈10 kcal/mol) activation energy, so we have not considered this direction, although it is known that nitration (especially in industry) has phenols as byproducts.
* Heats of formation of pre-reaction complexes were around 6–10 kcal/mol, so all found TSs were really the saddle points
* In each point geometry of supermolecule was optimized. Because the benzoic ring underwent slight deformation, the plane was drawn via its 1, 3, and 5 C-atoms.
*** In cases of substrates with low reactivity (nitrobenzene, pyridine) TSs were found to be “late.”