Новые аспекты механизма электрофильного ароматического замещения: квантово-химическая модель реакции нитрования
Газовая фаза
На рисунке 1 представлены возможные стадии реакции и стационарные точки, исследованные нами квантово-химически. Особенностью наших результатов при изучении газофазного процесса было обнаружение свободного предреакционного комплекса NO2+...C6H6. Этому состоянию соответствует большая область поверхности потенциальной энергии (ППЭ). NO2+ располагается над ароматическим кольцом на расстоянии примерно 3.3–3.5 Å без особых предпочтений в ориентации. Похоже, что этот комплекс можно классифицировать как сольватный комплекс. В пользу этого говорит факт, что нитроний координируется с О-содержащими бензольными производными (-OCH3, -COOH, -NO2 и т.д.) около атомов кислорода, а не π-системы. В таких случаях вопрос конформации предреакционного комплекса неоднозначен, поэтому все представленные ниже энтальпии рассчитаны относительно энтальпии образования изолированных исходных реагентов. Единственное неудобство в том, что для реакционноспособных аренов эффективная энергия активации становится формально отрицательной*. С другой стороны, известный экспериментальный факт, что в таких газофазных реакциях может наблюдаться отрицательная температурная зависимость у констант скорости [9].
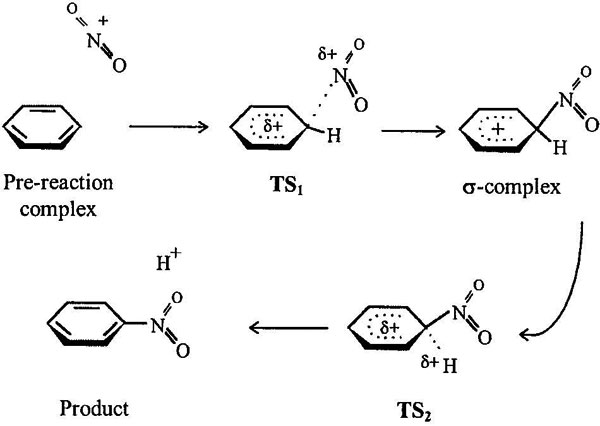 Figure 1. Возможнве стационарные точки на ППЭ.
Figure 1. Возможнве стационарные точки на ППЭ.
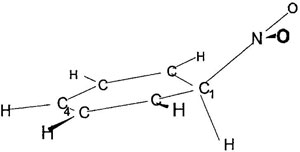 Figure 2. Геометрия σ-комплекса.
Figure 2. Геометрия σ-комплекса.
Упомянутый “сольватный” комплекс мог бы быть соотнесен с существующей в химии концепцией π-комплекса, если бы не речь шла не о газофазных расчетах. Забегая вперед сообщаем – в реакции в жидкой фазе нет никакого предреакционного комплекса, реагенты уже сольватированы (см. ниже раздел “Жидкая фаза”).
σ-Комплекс
Классическая концепция реакции нитрования ароматических соединений подразумевает взаимодействие положительно-заряженного атома азота нитрония с π-орбиталью углеродного атома бензольного кольца. Такое направление реакции ведет к аддукту, так называемому комплексу Уэланда (Wheland) или “сигма-комплексу” [3]. Наши расчеты дают для него длину C−N связи 1.56 Å. Бензольное кольцо слегка деформировано, диэдральные углы составляют 4–8° (см. Рис. 2). Судя по геометрии реакционного центра, он находится в состоянии, промежуточном между sp3 и sp2 гибридизацией. Связь C−H слегка ослаблена (растянута на 4%, а частота валентных колебаний составляет 2800 см−1 по сравнению с ≈3100 для остальных C−H колебаний). В комплексе наблюдается значительное перераспределение положительного заряда между нитрогруппой и субстратом. Положительный заряд делокализован между H-атомами (0.12–0.08, в зависимости от их удаленности от реакционного центра), а также реакционным центром, атомом углерода (0.3); нитрогруппа становится практически нейтральной.
Дальнейшее превращение, депротонирование, термодинамически не выгодно для газовой фазы и требует нуклеофила для формирования окончательного продукта, нитросоединения. Существует и измер для σ-комплекса, O-протонированный нитробензол. Термодинамически он более выгоден, на 30 ккал/моль, но синхронный механизм присоединения NO2+ и переноса протона требует очень напряженной структуры ПС (прямоугольник) с расстояниями C...H и O...H значительно бóльшими, чем естественные длины связей.
Присутствие в бензольном кольце заместителя создает возможность четырех различных направлений реакции присоединения электрофила: пара, мета, орто и ипсо атаки. Ипсо атака (атака на замещенный атом углерода) изучалась нами для оценки возможности существования ипсо-σ-комплекса как промежуточного продукта реакции. Орто направление имеет свою особенность: компланарная ориентация NO2+ становится невозможной из-за стерических препятствий (а объемные группы типа трет-бутила требуют полностью перпендикулярной ориентации реагентов). Это должно вести не только к увеличению энергии активации, но и к уменьшению энтропии активации реакции (и снижению констант скорости реакции).
ППЭ и путь реакции
Образованию σ-комплекса предшествует ПС. Обсудим возможные геометрии ПС. Сушествует широко распространенное мнение, что электрофилы дают с ароматическими соединениями π-комплексы, что подразумевает координацию электрофила над бензольным кольцом или одной из его π-связей. Мы построили ППЭ для исследования возможности такой координации и предреакционной селективности. На рисунках представлены сечения ППЭ описывающие энергию супермолекулы при размещении нитрония на расстоянии 3 Å над бензольным кольцом в параллельной плоскости**. Это расстояние соответствует начальному участку пути реакции (сравни: предреакционный комплекс 3.5 Å, ПС 2 Å). Такое сечение ППЭ для бензола выглядит как горка со впадинами вдоль C-H связей (Рис. 3). Таким образом, атака на центр кольца или C–C связей ведет не к притяжению, а сталкаванию электрофила к ближайшему атому углерода. Итак, мы не обнаружили ничего похожего на трехцентровые ПС, возможность которых аннонсирована в [10].
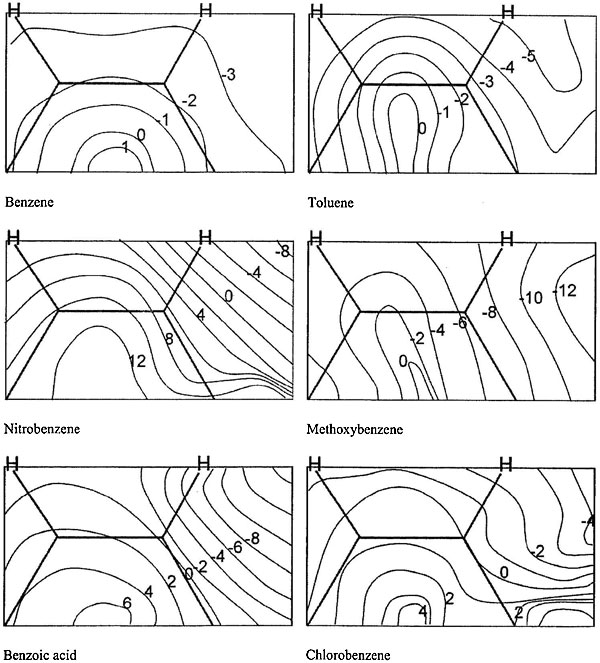 Рисунок 3. Сечения ППЭ, соответствующие расположению
Рисунок 3. Сечения ППЭ, соответствующие расположению NO2+ над ароматическим кольцом на расстоянии 3 Å. Показаны значения энергии относительно энтальпии образования реагентов (ккал/моль). Геометрии и ориентации реагентов оптимизированы в каждой точке. Группа заместителя расположена у крайнего справа C-атома.
Появление заместителя в ароматическом кольце деформирует и сдвигает этот энергетический холм на ППЭ от центра. Почти во всех случаях наблюдается понижение энергии в области над заместителем, что вызвано электростатическим притяжением. Особенно выраженный характер оно имеет в случаях О-содержащих заместителей, что может служить дополнительным фактором для орто-ориентации в реакции.
Особый предмет обсуждения механизма данной реакции – возможность переноса электрона. Существует мнение, основанное на экспериментальных данных, что NO2+ отнимает электрон от субстрата на начальном этапе реакции и дает ион-радикальную пару, которая на следующем этапе рекомбинирует и дает конечный продукт реакции [8]. Следует провести четкую границу в терминологии и различать перенос электронной плотности и собственно перенос электрона. Во избежание возможной путаницы, далее первый вариант мы будем именовать "перераспределение электронной плотности". Индикатором второго случая является не большое изменение атомных зарядов, а появление бирадикального характера супермолекулы. Мы рассмотрим данный вопрос подробнее в разделе , озаглавленном “Перенос заряда и перенос электрона”.
Переходные состояния (ПС)
Авторы [10] (3-21G, MP4) не нашли ПС газофазной реакции нитрония и бензола. Наоборот, наши расчеты практически для всех субстратов дают ПС со следующими характеристиками: угол атаки ≈120°, расстояние C−N 1.97–2.22 Å. Бензольное кольцо сохраняет свою плоскую конформацию. Нитрогруппа изогнута на 145°. ПС достигается как при компланарной, так и перпендикулярной ориентации плоскостей реагентов, соответствующие барьеры активации различаются менее чем на 1 ккал/моль, исключая случай орто-атак. Координатой реакции является практически чистая трансляция NO2+; никакого вклада колебаний C-H в координату реакции не обнаружено. Это означает, что ПС реакции нитрования в газовой фазе является "ранним" ***. В Таблице III кратко суммируется информация о характеристических точках пути реакции.
C1-N | ∠C4C1N | C1-H | ∠C4C1H | qNO2 | qN | qH | qC6H5 | ΔΔHf | |
|---|---|---|---|---|---|---|---|---|---|
| Изолированные реагенты | ∞ | – | 1.100 | 180 | 1.000 | 0.679 | 0.130 | −0.130 | 0 |
| Предреакционный комплекс | 3.3–3.5 | – | ∼1.10 | ∼180 | 1.000 | 0.684 | 0.155 | −0.155 | −6.1 |
| ПС1 | 2.071 | 118 | 1.115 | 151 | 0.452 | 0.609 | 0.202 | 0.347 | 3.9 |
| σ-комплекс | 1.559 | 144 | 1.156 | 112 | 0.026 | 0.564 | 0.252 | 0.723 | −9.3 |
| Продукт(ы) | 1.487 | 180 | ∞ | – | −0.150 | 0.567 | 1.000 | 0.150 | 97.1 |
Проведенные квантово-химические расчеты показали также возможность реакции между протонированным субстратом и молекулой нейтральной азотной кислоты: H-Ar-H+ + HO-NO2 ⇒ [H-Ar-NO2]+ + H2O. Реакция имеет практически тот же активационный барьер, хотя и требует особой ориентации реагентов для синхронного выброса H2O: N-OH связь молекулы азотной кислоты должна расположиться над HC-CH2 связью бензольного кольца. Следует упомянуть и еще одну возможность присоединения нитрония: атака атомом кислорода. Этот путь имеет более высокий барьер активации (≈10 ккал/моль), поэтому мы его специально не изучали, хотя известно, что при нитровании (особенно промышленном) в качестве побочных продуктов образуются фенолы.
* Энтальпии образования предреакционного комплекса составляли 6–10 ккал/моль, следовательно все наши ПС были действительно седловыми точками ППЭ.
** Геометрия супермолекулы оптимизировалась в каждой точке. Т.к. бензольное кольцо подвергалось некоторой деформации, плоскость проводилась через 1, 3 и 5 C-атомы.
*** В случаях субстратов с низкой реакционной способностью (нитробензол, пиридин) ПС были "поздние".