New aspects of electrophylic aromatic substitution mechanism: Computational model of nitration reaction
Liquid Phase
The SM2.1 calculations give many conclusions similar to those of gas phase. But there are certain differences, one example being that the process of solvation/desolvation of reagents results in classic profile of reaction with distinct activation barrier. Appearance of the barrier is due to positive-charge delocalization in TS caused by desolvation of NO2+ cation. The SM2.1 calculations predict no prereaction complex, meaning that there is no special affinity between NO2+ and aromatics, just solvation. The σ-complex becomes endothermic, unstable structure, in good agreement with chemical intuition. Its decomposition with H+-elimination is an exothermic process with hardly noticeable TS.
σ-Complex
In comparison with results of gas-phase calculations, σ-complex in liquid phase is characterized by more deformed and polarized C-H bond. It is lengthened by 15%, charge of H-atom is 0.43, and frequency of valent C-H vibration changes to 2200 sm−1 (from the usual 3100 sm−1). Reactions of low reactive substrates with late TS have no σ-complex at all: proton leaves synchronously with addition of NO2+.
Transition states and reaction coordinate
In the area of the first TS the reaction coordinate is again the length of C-N distance. The second stage of reaction, proton elimination, has more complex movement, including both valent and deformation vibrations of C-H bond. This second stage has practically no barrier, 1.5 kcal/mol for reaction of benzene (see Table IV). Traveling from TS along gradient vector, one can trace changes of energy or charges occurring during reaction (Figs. 4 and 5). Some parameters of stationary points on the pathway are shown in Table IV.
C1-N | ∠C4C1N | C1-H | ∠C4C1H | qNO2 | qN | qH | qC6H5 | ΔΔHf | |
|---|---|---|---|---|---|---|---|---|---|
| Isolated reagents | ∞ | — | 1.101 | 180 | 1.000 | 0.778 | 0.148 | −0.148 | 0 |
| TS1 | 1.977 | 122 | 1.124 | 144 | 0.445 | 0.644 | 0.229 | 0.326 | 42.7 |
| σ-complex | 1.537 | 148 | 1.277 | 103 | −0.159 | 0.512 | 0.432 | 0.727 | 22.9 |
| TS2 | 1.519 | 147 | 1.453 | 110 | −0.186 | 0.523 | 0.656 | 0.531 | 24.4 |
| Product | 1.484 | 180 | ∞ | — | −0.221 | 0.519 | 1.000 | 0.221 | −71.7 |
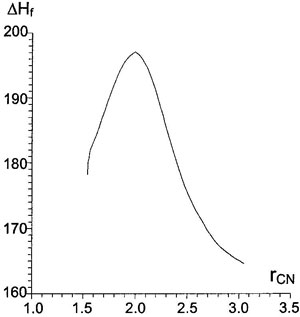 Figure 4. Dependence of heat of formation (kcal/mol) of supermolecule on
Figure 4. Dependence of heat of formation (kcal/mol) of supermolecule on C-N distance (Å) at the first stage (σ-complex formation) for AM1/SM2.1.
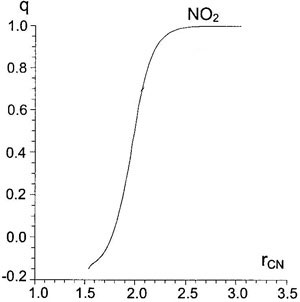 Figure 5. Dependence of charge of
Figure 5. Dependence of charge of NO2-group on C-N distance (Å) at first stage for AM1/SM2.1.