Radical addition to the vinyl C=C bond: Quantum chemistry model of the reaction
Structure of TSs and Intrinsic Reaction Coordinate of Radical Addition
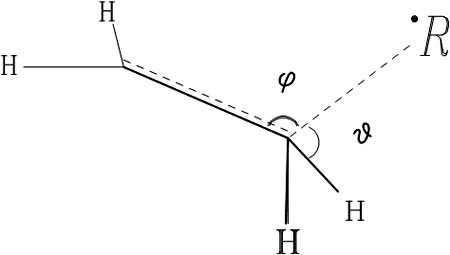
First, let us pay attention to the geometry and electronic structure of TSs of the considered reaction. Detailed investigation of reactions of ethene and chosen radicals was performed. Reagents approach each other in a coplanar “head-to-tail” manner. Depending on the radical the angle φ of attack is 100–105°; torsion angles θ of CH bond deviation are 10–15°, with valence angles ∠CH being≈120°.
 Reaction path was restored by gradient discern to both sides from the saddle point. The intrinsic reaction coordinate (IRC) appeared to be a complex of transition movement, umbrella vibration of
Reaction path was restored by gradient discern to both sides from the saddle point. The intrinsic reaction coordinate (IRC) appeared to be a complex of transition movement, umbrella vibration of =CH2, and, partly, valent vibration of C=C. When reaction passes its TS, the IRC gradually transforms into valent vibration of a newly formed bond. Analysis of energy and charge transformations along the reaction path could be summed shortly as follows (using HOO• and ethene as an example):
| 1 | R > 2.8 Å | Initial interaction, Coulomb interaction alone |
| 2 | R = 2.5 Å | Beginning of deformation of reaction center =CH2, π-bond unpairing, coupling of formed free valence of monomer with that of radical |
| 3 | R = 1.95 Å | TS (maximum of total energy) |
| 4 | R = 1.7 Å | Beginning of “chemical bond” formation, corresponding IRC vibration becomes real; resonance part of total energy achieves maximum due to orbitals' unbonding; maximum of charge separation |
| 5 | R = 1.43 Å | Product |