Radical addition to the vinyl C=C bond: Quantum chemistry model of the reaction
Results of the PMO Theory Approach
The PMO theory remains the best tool to discuss the reactivity of various chemical reactions. Our reaction is a suitable case for its application and, moreover, in π-approximation. The interaction energy of the PMO theory, also called «charge transfer» (CT) energy, has a well-known form:
(1)
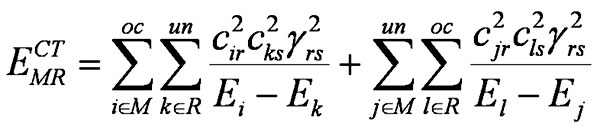
where summing is done over a set of spin orbitals and γ is the resonance integral on the atomic orbitals (AOs) of reactive centers r and s.
Using RHF π-orbitals of isolated reagents we calculated CT energies of all series in consideration (see, e.g., Tables V and VI). Figure 4 shows that there is some symbasis between them and the activation barriers of corresponding reactions. Addition to a substituted position gives less satisfactory correlations that are obviously caused by a greater steric effect (deformation of reaction center). So, ECT could also be taken for another factor of reactivity. But, there is one inconvenience with it: It is not an ordinary index of reactivity; it is the whole set of energies of MOs and their coefficients of the AOs of the reaction center. It is no wonder that when using the PMO theory researchers frequently truncate orbital sets to HOMO and LUMO to avoid the summing in Eq. (1); this method is known as the frontier molecular orbitals (FMO) approach.
| Monomer | β-attack | α-attack | ||||||
|---|---|---|---|---|---|---|---|---|
| Ea | ECT | N | G | Ea | ECT | N | G | |
| Ea, kcal/mol; ECT, (γ2/eV); N and G, in eV. | ||||||||
| Eth | 26.4 | −0.286 | 1.304 | 14.1 | ||||
| Prop | 25.8 | −0.294 | 1.147 | 13.7 | 31.0 | −0.285 | 1.460 | 14.2 |
| AN | 26.0 | −0.287 | −1.740 | 14.1 | 30.3 | −0.271 | 0.284 | 14.8 |
| MA | 26.8 | −0.284 | −2.187 | 14.4 | 31.3 | −0.270 | 0.726 | 14.8 |
| VA | 23.9 | −0.307 | 2.011 | 13.3 | 26.8 | −0.278 | 0.738 | 14.4 |
| VCl | 26.5 | −0.284 | −0.355 | 14.1 | 30.2 | −0.273 | 0.233 | 14.7 |
| VCl2 | 25.7 | −0.289 | −1.625 | 14.0 | 32.0 | −0.266 | −0.459 | 15.1 |
| St | 23.0 | −0.311 | 1.371 | 13.0 | ||||
| ViNaph | 22.7 | −0.313 | 1.379 | 12.9 | ||||
| 2ViPy | 23.3 | −0.303 | 0.788 | 13.3 | ||||
| 4ViPy | 23.4 | −0.299 | 0.125 | 13.4 | ||||
| Bu2ene | 22.6 | −0.310 | 1.395 | 13.1 | ||||
| Hex3ene | 21.8 | −0.315 | 1.400 | 12.9 | ||||
| Monomer | β-attack | α-attack | ||||||
|---|---|---|---|---|---|---|---|---|
| Ea | ECT | N | G | Ea | ECT | N | G | |
| Ea, kcal/mol; ECT, (γ2/eV); N and G, in eV. | ||||||||
| Eth | 13.9 | −0.349 | −0.735 | 11.5 | ||||
| Prop | 13.6 | −0.359 | −0.960 | 11.2 | 18.1 | −0.348 | −0.675 | 11.5 |
| St | 11.4 | −0.378 | −1.054 | 10.7 | −0.344 | −0.766 | 11.7 | |
| AN | 13.0 | −0.391 | −3.405 | 11.3 | 17.0 | −0.355 | −1.714 | 11.5 |
| MA | 13.4 | −0.388 | −3.738 | 11.5 | 18.0 | −0.349 | −1.342 | 11.6 |
| VA | 12.1 | −0.370 | −0.385 | 10.8 | 16.7 | −0.346 | −1.353 | 11.7 |
| VCl | 13.3 | −0.369 | −2.216 | 11.3 | 16.9 | −0.354 | −1.716 | 11.5 |
| VCl2 | 12.6 | −0.396 | −3.343 | 11.1 | 19.6 | −0.363 | −2.394 | 11.5 |
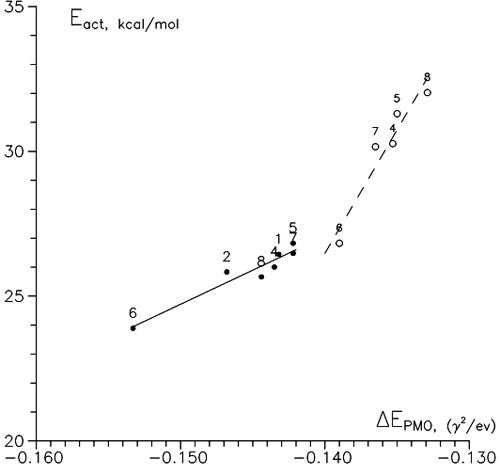 Figure 4. Relationships between activation energies and CT energy of the PMO method,
Figure 4. Relationships between activation energies and CT energy of the PMO method, HOO addition. •, β-attack; ○, α-attack.
C=Cbond (both experimental and calculational data) appears to be completely inert from the point of view of the FMO approach. Obviously, this is due to truncation of many π-orbitals in this case, not to the different electronic structure of styrene.