Radical addition to the vinyl C=C bond: Quantum chemistry model of the reaction
Empirical Schemes of Vinyl Monomers' Relative Reactivity in Radical Addition Reactions
In the previous paragraph we proposed to try Eq. (3), or just its polar part, as a basis of nonlinear empirical schemes of reactivity. This approach is intended for calculation or prediction of rate constants (or activation barriers) in cases when the differences in the structure of reagents are too great to use linear correlation schemes of the Hammett type. Let us try to build a scheme of reactivity of vinyl monomers in radical addition reactions. Firstly, we can use only the form of Eq. (3), its parameters being the variables for the optimization procedure. On the other hand, we can try to use theoretically obtained parameters of vinyl monomers as indices of reactivity and correlate experimental data with them.
Both variants were studied. Let's start from entirely empirical method, the first approach. For this purpose we are going to use a slightly simplified form of Eq. (3) (supposing only EN influences the value of CT energy(7) ):
(4)
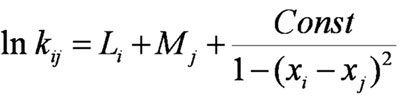
where Li and xi are parameters of a radical and Mj and xj of a monomer (x is the EN of a reagent; L and M collect entropy, nonpolar effects, and so on). Because only relative rate constants of radical addition to monomers were considered (ln kij/kik), parameter L never appeared. Parameterization of the scheme was accomplished on the basis of approximately 200 rate constants [1, 7, 9] of reactions between 20 chosen radicals and 20 vinyl monomers. All obtained parameters are shown in Table IX (see more data in Ref. [10]). Comparison of calculated values with experimental data are shown in Figure 7. It enables us to say that the proposed scheme describes these experimental data reasonably well (mean deviation is less than 50%, the precision of those kinetic data themselves). So, the form of Eq. (4) is both meaningful and adequate for the purposes of nonlinear correlation tasks. It is relevant to add here that all parameters also “correlate” with common chemical views on donor–acceptor properties of both the radicals and monomers that were considered.
| Monomer | M | x | Radical | x |
|---|---|---|---|---|
* Styrene parameters were used as reper values. ** Additional abbreviations of vinyl monomers: MSt, α-methylstyrene; MAN, methacrylic nitryl; MMA, methyl methacrylate; IPA, isopropenylacetate; VBuE, vinyl buthyl ether; ViPy, vinylpyridine; MVK, methylvinylketone; DPE, diphenylethene; VF, vinyl fluoride. *** Polyalkyl radicals | ||||
| St | 0* | 0 | CH3• | 0.477 |
| MSt** | 0.284 | 0.0081 | t-Bu | 0.518 |
| AN | −1.908 | −0.0559 | c-C6H11• | 0.587 |
| MAN | −1.585 | −0.0218 | 5-C6H9• | 0.528 |
| MA | −2.085 | −0.0482 | C6H5• | 0.310 |
| MMA | −1.006 | −0.0315 | C6H5-S• | −0.534 |
| VA | −4.582 | −0.0285 | t-BuO• | −0.482 |
| IPA | −4.061 | −0.0149 | t-BuOO• | −0.541 |
| VBuE | −5.200 | 0.0102 | PhC(CH3)2OO• | −0.372 |
| 2-ViPy | 0.251 | −0.0122 | CCl3• | −0.204 |
| 4-ViPy | −0.023 | −0.0114 | CF3• | −0.627 |
| 1,1-DPE | 1.216 | 0.0122 | ||
| 1,2-DPE | −1.660 | 0.0093 | poly-St*** | 0.446 |
| VCl | −3.034 | −0.0253 | poly-MSt | 0.598 |
| VCl2 | −2.097 | −0.0449 | poly-AN | −0.293 |
| VF | −5.087 | −0.0219 | poly-MAN | 0.072 |
| VF2 | −3.522 | −0.0604 | poly-MA | 0.157 |
| Eth | −4.431 | −0.0167 | poly-MMA | 0.088 |
| MVK | −1.729 | −0.0561 | poly-VA | 0.067 |
There are experimental data on reactivity of radicals and vinyl monomers that have substituted benzoic ring in theirs structure. In the process of parametrization we assumed that substituent in such aromatic compounds only shifts EN of the reagent: xi=x0 + ρ σi, where ρ is the sensitivity quotient and σi is parameter of substituent (they correspond both by the meaning and value the Hammett's ρ/σ parameters [2]); see Table X.
| substituent | σ** | group | ρ |
|---|---|---|---|
| *gained by optimization procedure; **σH = 0; ***ρPhS = 1 was set as the reper value | |||
p-NH2 | 0.111 | Ph-S • | 1.0*** |
p-OCH3 | 0.042 | PhCH=CH2 | 0.145 |
p-CH3 | 0.004 | PhCH •∼ | 0.936 |
p-Cl | −0.013 | Ph• | 2.4 |
p-Br | −0.009 | ||
p-CN | −0.030 | ||
p-NO2 | −0.041 | ||
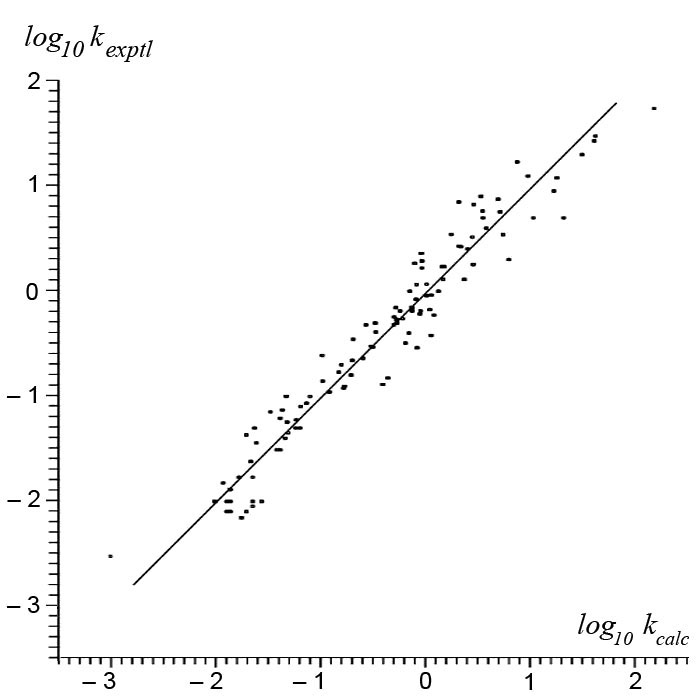 Figure 7. Relationship between experimental relative rate constants and calculated values [Eq. (4)]
Figure 7. Relationship between experimental relative rate constants and calculated values [Eq. (4)]
Another method of utilizing Eq. (3) is using its polar part as a theoretical index of reactivity. So, we are going to tabulate electronegativities and energy gaps of monomers and use them as parameters of activity for correlation purposes. This time only linear (Mj) parameters of monomers shall be obtained through optimization. The working equation of such a scheme be
(5)
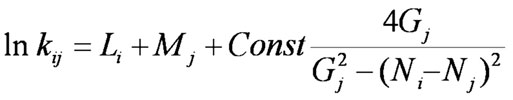
The set of optimized parameters Mj is presented in Table XI. The quality of prediction of this scheme is a bit lower than that of the previous one (mean deviation of calculated rate constants is approximately 60%). But, it has the considerable advantage of being “semiempirical” because the number of empirical parameters is essentially reduced and an important part of predictions is based on values calculated by quantum chemistry.
| Monomer | M | N | G | Radical | N |
|---|---|---|---|---|---|
Parameters N/G of monomers calculated with relatively HOO• were used. Parameters N of radicals were obtained by optimization procedure, while G of different reaction series were supposed to be equal. | |||||
| St | 0.00 | 1.37 | 13.0 | CH3• | 1.26 |
| MSt | −0.45 | 1.19 | 12.8 | c-C6H11• | 2.75 |
| AN | 0.78 | −1.74 | 14.1 | 5-C6H9• | 3.42 |
| MAN | 1.05 | −1.69 | 13.8 | t-Bu• | 2.52 |
| MA | 0.76 | −2.19 | 14.4 | t-BuO• | −2.71 |
| MMA | 1.39 | −2.08 | 14.0 | t-BuOO• | −2.74 |
| VA | −4.42 | 2.01 | 13.3 | PhC(CH3)2OO• | −2.36 |
| IPA | −5.50 | 2.00 | 13.0 | Cl-PhS• | −2.73 |
| Eth | −2.04 | 1.30 | 14.1 | CF3 | −4.00 |
| VBuE | −6.34 | 3.19 | 13.2 | CCl3 | −0.70 |
| VCl | −0.73 | −0.36 | 14.1 | poly-IPA | −0.76 |
| VCl2 | 0.55 | −1.63 | 14.0 | poly-VA | −0.82 |
| 2ViPy | 0.80 | 0.79 | 13.3 | poly-MMA | −1.03 |
| 4ViPy | 0.87 | 0.13 | 13.4 | poly-MA | −0.52 |
| MVK | 0.86 | −1.46 | 14.2 | poly-MAN | −1.43 |
| 1,1-DPE | 2.86 | 0.80 | 13.6 | poly-AN | −2.09 |
| VF | −3.28 | 1.00 | 13.7 | poly-MSt | 2.73 |
| poly-St | 1.86 | ||||
(7) The parameter corresponding to the energy gap of reagents is absent in this scheme because it has been found that it may be set constant (variation of such an expanded form of equation gave no improvement).