Radical addition to the vinyl C=C bond: Quantum chemistry model of the reaction
Comparison of Direct TS Calculations with Indices of Reactivity
Let us return to our consideration of radical addition. Earlier, we suggested that the activation energy of this reaction is a total of two effects: deformation and polar interaction. The first can be evaluated by standard deformation energy, E0def (see above), and the second by CT energy of the PMO method. To try the possibility of a general scheme of radical activity, all previously obtained results of direct TS calculations were considered within the following equation:
(3)
Ea = a0 + a1E0def + a2ECT
To use the advantages of the N/G method of calculation of ECT we used the formula (2')
![]()
Parameters N and G calculated relatively HOO• were used for the standard set of monomers' parameters. Parameters of radicals could be easily derived from linear dependencies shown in Figure 5. So, we just needed to obtain parameters a0, a1, a2 of reactions. That was done by the usual optimization procedure. But, it was found that parameters of radicals obtained from π-approximation poorly correspond to the true picture of reactivity (and, really, they have considerable σ-character in TS). So, we decided to adjust parameters of radicals within the same optimization process. Final optimized values are collected in Tables VII and VIII. Figure 6 shows the quality of usage of Eq. (3) for evaluation of activation energies.
| β-addition | α-addition | |||||
|---|---|---|---|---|---|---|
| Edef | N | G | Edef | N | G | |
| Edef, kcal/mol; N, G, eV | ||||||
| Eth | 6.83 | 1.30 | 14.1 | |||
| Prop | 6.51 | 1.15 | 13.7 | 7.45 | 1.46 | 14.2 |
| AN | 6.57 | −1.74 | 14.1 | 7.19 | 0.28 | 14.8 |
| MA | 7.01 | −2.19 | 14.4 | 7.65 | 0.73 | 14.8 |
| VA | 5.64 | 2.01 | 13.3 | 6.10 | 0.74 | 14.4 |
| VCl | 6.56 | −0.36 | 14.1 | 7.08 | 0.23 | 14.7 |
| VCl2 | 6.07 | −1.63 | 14.0 | 6.96 | −0.46 | 15.1 |
| St | 4.82 | 1.37 | 13.0 | 7.88 | 1.40 | 14.3 |
| Bu2 | 4.786 | 1.39 | 13.1 | |||
| Hex3 | 4.438 | 1.40 | 12.9 | |||
| ViNaph | 4.890 | 1.38 | 12.9 | |||
| 2ViPy | 5.204 | 0.79 | 13.3 | |||
| 4ViPy | 5.113 | 0.12 | 13.4 | |||
| 4ViPm | 5.611 | −0.42 | 13.6 | |||
| 2ViPm | 5.783 | −0.32 | 13.4 | |||
| N | G | β-addition | α-addition | |||
|---|---|---|---|---|---|---|
| a0 | a1 | a0 | a1 | |||
* HOO is chosen to be the reper reactant, so relative parameters of other radicals are presented here (N < 0 means a more electronegative and G < 0 a more “sensitive” radical). | ||||||
HOO | 0 | 0 | 28.0 | 1.46 | 20.8 | 2.85 |
CH3 | 1.60 | −1.00 | 21.0 | 0.75 | 23.8 | 0.75 |
CH3O | −0.55 | −0.65 | 26.2 | 0.92 | 14.0 | 3.05 |
CCl3 | −1.75 | −1.30 | 28.6 | 0.80 | 24.3 | 2.00 |
CF3 | −3.50 | −0.23 | 23.8 | 0.60 | 16.0 | 2.30 |
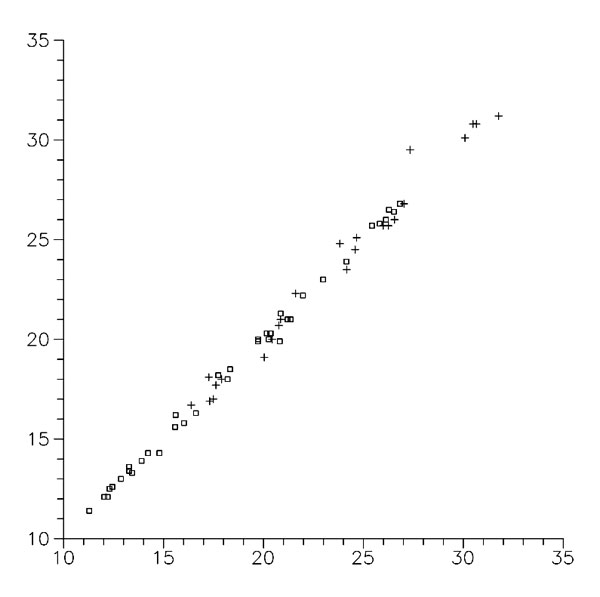 Figure 6. Relationship between directly calculated activation energies and those evaluated by Eq. (3) values, kcal/M (β-addition indicated by □, α-addition by +).
Figure 6. Relationship between directly calculated activation energies and those evaluated by Eq. (3) values, kcal/M (β-addition indicated by □, α-addition by +).
The main significance of that correlation is that regulations of radical addition to the C=C bond (how they are seen by semiempirical methods of quantum chemistry) could be explained by the electronic structure of reagents using two indices of reactivity: ability of reaction center to deform and change its hybridization and the energy of interaction of π-MOs of reagents. It is a justification of our next step: to explain experimental regulations by these same characteristics of reagents and try the mathematical form of Eq. (3) for a correlation analysis of experimental data.