Новые аспекты механизма электрофильного ароматического замещения: квантово-химическая модель реакции нитрования
Анализ реакционной способности субстратов
Термохимический аспект
Из-за сходства электронной структуры ПС1 и σ-комплекса мы ожидали проявление правила линейности свободных энергий на этой элементарной стадии реакции. Такая корреляция представлена на Рис. 6. (Отрицательные значения некоторых барьеров активации обсуждены выше, в разделе “Газовая фаза”). График показывает, что точки, соответствующие орто-атаке ложатся на линию, параллельную корреляционной прямой для пара и мета ориентаций. Очевидно, что это вызвано проявлением стерического взаимодействия (если быть точнее, стерические препятствия проявляются в σ-комплексе, увеличивая его энтальпию образования). Что до ипсо-присоединения – здесь корреляция не слишком хороша, опять из-за большого влияния стерического фактора. С другой стороны, такая же плохая корреляция и между барьерами активации и общей энтальпией реакции нитрования. Это можно объяснить отсутствием деформации бензольного кольца в конечных продуктах нитрования.
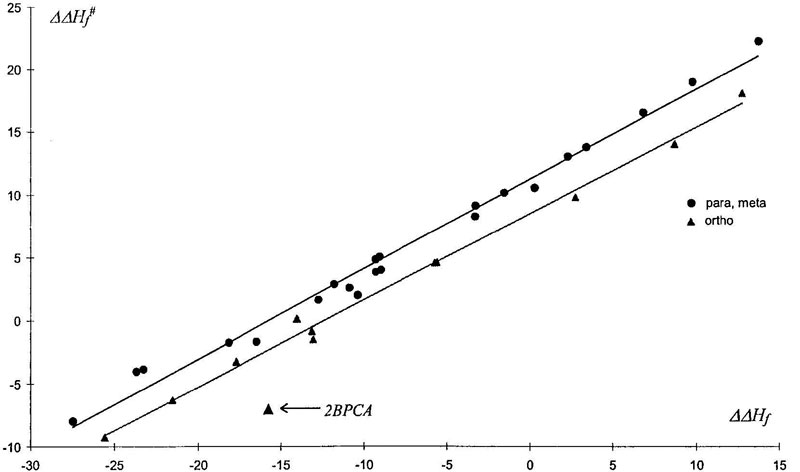 Figure 6. Соотношение между энергиями активации (ΔΔH≠f, ккал/моль) и энтальпиями образования σ-комплекса (ΔΔHf, ккал/моль).
Figure 6. Соотношение между энергиями активации (ΔΔH≠f, ккал/моль) и энтальпиями образования σ-комплекса (ΔΔHf, ккал/моль).
Так что в качестве индекса реакционной способности может послужить лишь энтальпия образования σ-комплекса, но не общая теплота реакции.
Интересный случай орто-эффекта был обнаружен в реакции с 2-бифенилкарбоновой кислотой (2BPCA). Геометрия этой молекулы такова, что в ПС карбоксильная группа развернута отрицательно заряженными атомами кислорода в сторону атакующего положительно заряженного электрофила. Это дает значительный выигрыш в энергии ПС (2 ккал/моль для орто-атаки и 1 ккал/моль для пара)*. Такое дополнительное взаимодействие существует только в ПС, т. к. в σ-комплексе заряд нитрогруппы практически равен нулю; что и приводит к большому отклонению от корреляционной прямой (см. Рис. 6).
У нас есть возможность сравнить полученные в результате расчета барьеры активации с экспериментально наблюдаемой активностью субстратов. Attina и Cacace [13] измерили константы скорости реакции CH3O-NO2+ с алкил- и галоген- бензолами в газовой фазе. Рис. 7 показывает, что расчеты находятся в хорошем согласии с наблюдаемой реакционной способностью данных соединений. Расчеты термохимии методом SM2.1 дают аналогичные результаты: наблюдается линейная корреляция между энергией активации и энтальпией образования σ-комплекса. Соответствующие результаты собраны в Таблице VII.
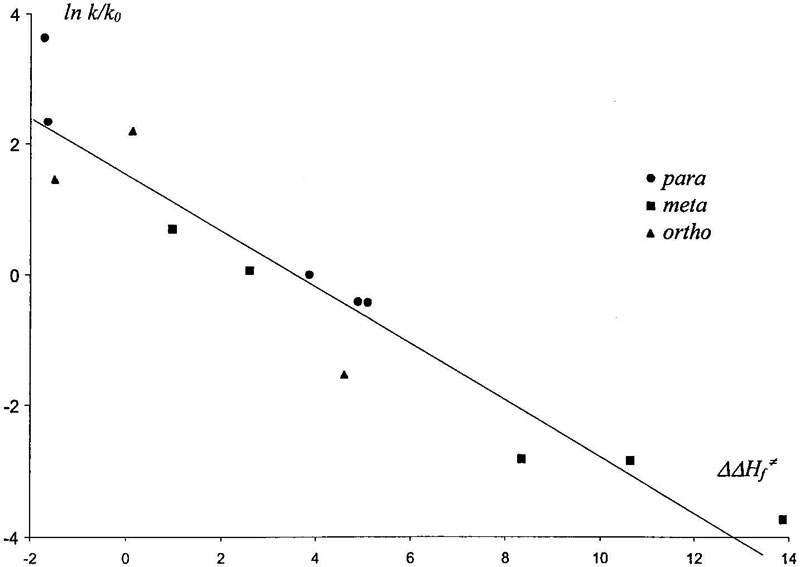 Figure 7. Логарифмическая зависимость экспериментальной реакционной способности от расчетных энергий активации. Парциальные константы скорости газофазной реакции
Figure 7. Логарифмическая зависимость экспериментальной реакционной способности от расчетных энергий активации. Парциальные константы скорости газофазной реакции CH3O-NO2+ с метилбензолом, трет-бутилбензолом, хлор- и фтор- бензоламирассчитаны относительно константы скорости присоединения к бензолу по данным [13].
| X | ΔΔH≠f | ΔΔHσf | ΔΔHf |
|---|---|---|---|
| * σ-комплекс не существует | |||
H | 42.74 | 22.84 | −71.74 |
CH3 | |||
| пара | 40.67 | 20.48 | −72.04 |
| мета | 43.28 | 24.06 | −71.61 |
| орто | 41.93 | 24.33 | −69.71 |
| ипсо | 46.03 | 35.56 | – |
Ph | |||
| пара | 41.64 | 21.46 | −70.89 |
| мета | 44.22 | 25.62 | −70.68 |
| орто | 42.87 | 26.79 | −66.47 |
| ипсо | 50.45 | 38.22 | – |
OAc | |||
| пара | 41.83 | 21.59 | −69.99 |
| мета | 46.49 | 28.71 | −69.65 |
| орто | 44.23 | 26.34 | −68.39 |
| ипсо | 53.86 | 46.23 | – |
Cl | |||
| пара | 44.50 | 25.29 | −69.72 |
| мета | 46.63 | 28.48 | −69.29 |
| орто | 45.27 | 28.96 | −64.64 |
| ипсо | 52.46 | 40.03 | — |
F | |||
| пара | 42.76 | 23.00 | −69.70 |
| мета | 47.36 | 29.40 | −68.68 |
| орто | 43.71 | 24.98 | −68.86 |
COOH | |||
| пара | 49.81 | * | −67.76 |
| мета | 46.55 | 28.64 | −68.41 |
| орто | 50.33 | 34.38 | −63.65 |
| ипсо | 56.18 | 47.19 | — |
NO2 | |||
| пара | 54.44 | * | −65.31 |
| мета | 51.11 | * | −66.10 |
| орто | 54.79 | 39.79 | −62.15 |
| Пиридин, мета | 46.14 | 26.73 | −69.68 |
| Нафталин | |||
| α | 39.86 | 19.58 | −67.12 |
| β | 42.13 | 22.45 | −70.56 |
| Мезитилен | 40.77 | 21.84 | −67.55 |
| ипсо | 46.95 | 37.07 | – |
| Гексаметилбензол | 41.72 | 26.77 | – |
Процедура разделения энергии
Ea = ΔEA + ΔEB + EAB
Процедура разделения энергии на ее одно- и двухцентровые составляющие послужила нам скорее для качественных, чем количественных оценок. В большинстве случаев невозможно сделать определенных выводов на основе значений энергии стабилизации EAB, поскольку абсолютные цифры имеют обратную зависимость: чем больше активационный барьер (т.е. чем ниже реакционная способность), тем сильнее межмолекулярное взаимодействие. При анализе можно выделить деформационную часть ΔEA и ΔEB в добавок к энергии взаимодействия EAB. Анализ EAB позволил обнаружить еще один фактор реакционной способности субстратов: кулоновское взаимодействие электрофила и заместителя в кольце. Дело в том, что в ПС это взаимодействие значительно отличается по величине и знаку от начального, и полностью исчезает в σ-комплексе (из-за зарядовой нейтрализации нитрогруппы). Поэтому реальная активность может отличаться от предсказаний как на основе структуры реагентов, так и стабильности продуктов реакции. К примеру, Таблица VIII демонстрирует некоторые характеристики, полученные при анализе результатов процедуры разделения энергии. Следует обратить внимание на свидетельства обсуждаемого эффекта в реакциях фенилацетата и 2-бифенилкарбоновой кислоты (X = -O-COCH3 and -Ph-COOH).
| −X | Ea, kcal/mol | EAB, eV | Eres, eV | Eq, eV | QX, e |
|---|---|---|---|---|---|
| * Заряд NO2 группы в ПС ≈ 0.40 ÷ 0.45 e. | |||||
-CH3 | 0.1 | −2.503 | −2.386 | 0.268 | 0.157 |
-CF3 | 14.1 | −3.202 | −3.560 | 0.221 | 0.066 |
-C4H9, tret | −1.5 | −2.326 | −2.160 | 0.244 | 0.153 |
-NO2 | 18.2 | −3.351 | −3.748 | −0.016 | −0.026 |
-COOH | 9.9 | −3.228 | −3.442 | −0.032 | 0.017 |
-Cl | 4.6 | −2.679 | −2.662 | 0.231 | 0.129 |
-O-COCH3 | −6.3 | −2.888 | −2.547 | −0.279 | 0.039 |
-Ph | −4.0 | −2.748 | −2.358 | −0.004 | 0.247 |
-Ph-COOH, орто | −6.9 | −3.533 | −3.389 | −0.267 | 0.098 |
Теория возмущенных молекулярных орбиталей (ВМО), индекс реакционной способности
Молекцлярные орбитали реагентов, полученные в газофазном приближении, не пригодны для рассмотрения в рамках ВМО, так как EВЗМО (высшая занятая МО) арена и EНСМО (низшая свободная МО) нитрония или нитрацидия практически вырождены, их энергия −9 eV. Поэтому анализ проводили на основе МО, полученных в расчетах жидкофазного приближения, где электроотрицательности электрофилов существенно ниже: EНСМО ≈ −2 eV). Энергия взаимодействия реагентов, рассчитанная методом ВМО, дает неудовлетворительную корреляцию с расчетными энергиями активации соответствующих реакций, показывая, что межмолекулярное взаимодействие – не единственный фактор реакционной способности.
Вводя параметр, описывающий кулоновское взаимодействие (см. выражение в Таблице VIII), дает нам двухпараметровую линейную корреляционную схему. При этом коэффициент корреляции возрастает с 0.82 до 0.94, и тем не менее такая схема кажется неадекватной без учета фактора деформации. Энергию деформации можно оценить искривлением ароматического колца к некоторой конкретной геометрии. Но давайте рассмотрим другой, более естественный процесс: протонирование ароматического соединения. Все вышеупомянутые факторы (деформация реакционного центра, электростатическое взаимодействие, молекулярное взаимодействие) присутствуют в таком процессе и количественно соединяются в энтальпию протонирования. Как видно из Рис. 8, корреляция энергий активации нитрования с данным параметром даже лучше, чем с энтальпией образования σ-комплекса (Рис. 6). Объяснение заключается в том, что в σ-комплексе (продукте присоединения) NO2 группа уже практически не имеет заряда, в то время как в H+-комплексе присутствует значительное электростатическое взаимодействие между электрофилом (H+) и заместителем, так же, как и в переходном состоянии реакции нитрования. Кажется, это идеальный индекс реакционной способности субстратов для нитрования (и подобных электрофильных реакций), так как он дает единую общую корреляцию для орто, мета и пара замещения, и удовлетворительно покрывает случаи ипсо-замещения и особые случаи орто эффектов.
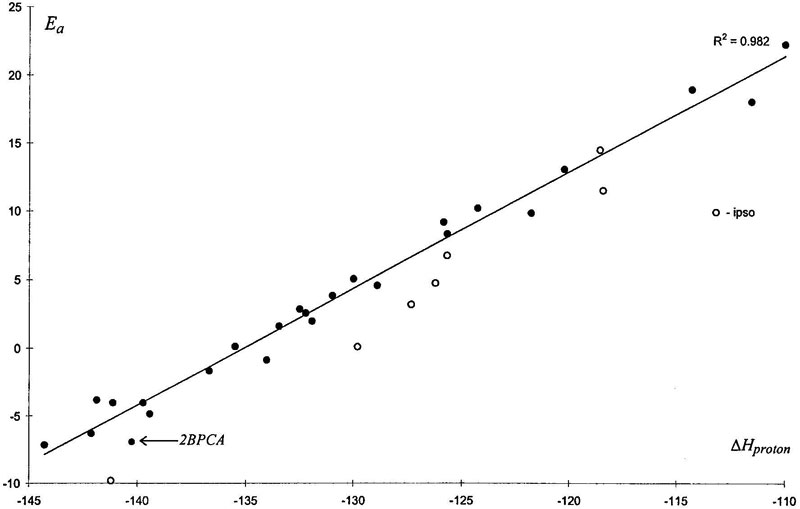 Рисунок 8. Зависимость энергий активации нитрования от энтальпии протонирования, AM1, газофазное приближение, ккал/моль (•, орто, мета и пара; ○, ипсо атака).
Рисунок 8. Зависимость энергий активации нитрования от энтальпии протонирования, AM1, газофазное приближение, ккал/моль (•, орто, мета и пара; ○, ипсо атака).
* Повышенная активность 2BPCA – экспериментальный факт, особенно высокое орто/пара соотношение продуктов реакции (от 3 до 8 в зависимости от условий реакции) [12].